

50年難遇AI“諾獎級”里程碑!DeepMind破解蛋白質(zhì)折疊難題
歡迎關(guān)注“創(chuàng)事記”的微信訂閱號:sinachuangshiji
文/新智元
來源:新智元(ID:AI_era)
原標題:50年難遇AI“諾獎級”里程碑!DeepMind破解蛋白質(zhì)折疊難題,Nature:這可能改變一切
剛剛,一個困擾生物學家50年的難題,被AI解決了。
去年年底,谷歌DeepMind推出了一種名為AlphaFold(一個用人工智能加速科學發(fā)現(xiàn)的系統(tǒng),它基于蛋白質(zhì)的基因序列,就能預測蛋白質(zhì)的3D結(jié)構(gòu))的算法。
今天,在有“蛋白質(zhì)奧林匹克競賽”稱呼的國際蛋白質(zhì)結(jié)構(gòu)預測競賽(CASP)上,AlphaFold擊敗了其余的參會選手,能夠精確地基于氨基酸序列,預測蛋白質(zhì)的3D結(jié)構(gòu)。
其準確性可以與使用冷凍電子顯微鏡(CryoEM)、核磁共振或 X 射線晶體學等實驗技術(shù)解析的3D結(jié)構(gòu)相媲美。
DeepMind聯(lián)合創(chuàng)始人及CEO德米斯·哈薩比斯(Demis Hassabis)表示:“DeepMind背后的終極愿景一直是構(gòu)建通用人工智能,利用通用人工智能來極大地加速科學發(fā)現(xiàn)的步伐,幫助我們更好地了解周圍世界。”

谷歌CEO桑達爾·皮查伊(Sundar Pichai)在Twitter上為此次突破點贊。
這是一個跨圈兒的壯舉,李飛飛、馬斯克等大佬也紛紛點贊:


《自然》雜志更是評論其為“這將改變一切”。

五十年來,蛋白質(zhì)折疊一直是生物學的巨大挑戰(zhàn)。
分子折疊方式變幻無窮,其重要性很難估計。大多數(shù)生物過程都圍繞蛋白質(zhì),而蛋白質(zhì)的形狀決定了其功能。只有當知道蛋白質(zhì)如何折疊時,他我們才能知曉蛋白質(zhì)的作用。
例如,胰島素如何控制血液中的糖水平以及抗體如何對抗冠狀病毒,都由蛋白質(zhì)的結(jié)構(gòu)來決定。

我們都知道,DeepMind以戰(zhàn)勝人類而著名,在國際象棋,圍棋,星際爭霸II和老式的Atari經(jīng)典游戲中都占據(jù)了上風。
但超人游戲從來都不是主要目標,游戲為程序提供了訓練場,一旦程序足夠強大,就可以解決現(xiàn)實世界中的問題!
蛋白質(zhì)折疊50年來重大突破,AI破解預測難題
蛋白質(zhì)的形狀與其功能密切相關(guān),預測蛋白質(zhì)結(jié)構(gòu)的能力可以幫助我們更好地理解蛋白質(zhì)的功能和工作原理。世界上許多重大的挑戰(zhàn),比如發(fā)展疾病的治療方法或者找到分解工業(yè)廢物的酶,從根本上來說都與蛋白質(zhì)及其所扮演的角色有關(guān)。
傳統(tǒng)上,得到蛋白質(zhì)的形狀需要花費數(shù)年的時間。
從1950年代開始,使用X射線束照射結(jié)晶的蛋白質(zhì)并將衍射光轉(zhuǎn)化為蛋白質(zhì)原子坐標的技術(shù),確定了蛋白質(zhì)的第一個完整結(jié)構(gòu)。X射線晶體學已經(jīng)證明了蛋白質(zhì)結(jié)構(gòu)的絕大部分。但是,在過去的十年中,低溫電子顯微鏡已成為許多結(jié)構(gòu)生物學實驗室的首選工具。

最新的技術(shù)進步已使使用低溫電子顯微鏡生成接近原子分辨率的電子密度圖成為可能
這些方法依賴于進行大量試驗和改進錯誤,可能需要花費數(shù)年的工作時間來完成每個蛋白質(zhì)結(jié)構(gòu),并需要使用數(shù)百萬美元的專門設(shè)備來進行試驗和驗證。
1969年,塞勒斯 · 萊文塔爾(Cyrus Levinthal)指出,用暴力計算法列舉一個典型蛋白質(zhì)的所有可能構(gòu)型所需的時間比已知宇宙的年齡還要長,他估計一個典型蛋白質(zhì)有10 ^ 300種可能構(gòu)型。
1972年,克里斯蒂安 · 安芬森(Christian Anfinsen)在諾貝爾化學獎的獲獎感言中,提出了一個著名的假設(shè):
 克里斯蒂安· 安芬森
克里斯蒂安· 安芬森理論上來說,蛋白質(zhì)的氨基酸序列應(yīng)該完全決定其結(jié)構(gòu)。
這個假設(shè)引起了長達五十年的探索,即僅僅基于蛋白質(zhì)的 1D 氨基酸序列就能夠計算預測蛋白質(zhì)的 3D 結(jié)構(gòu)。然而,這個假設(shè)面臨的一個主要的挑戰(zhàn)是,理論上蛋白質(zhì)在進入最終的 3D 結(jié)構(gòu)之前可以折疊的方式數(shù)量是一個天文數(shù)字。
1980年代和1990年代,盡管早期計算機科學家已經(jīng)取得了進展,但是從蛋白質(zhì)的組成中推斷結(jié)構(gòu)仍非易事。
為什么預測蛋白質(zhì)的形狀非常困難?
在自然界中,蛋白質(zhì)是氨基酸鏈,可以自發(fā)折疊成無數(shù)令人難以想象的形狀,有些甚至在幾毫秒之內(nèi)完成。
為了了解蛋白質(zhì)如何折疊,DeepMind的研究人員在一個包含約170,000個蛋白質(zhì)序列及其形狀的公共數(shù)據(jù)庫中對其算法進行了訓練。在相當于100到200個圖形處理單元(按現(xiàn)代標準,計算能力適中)上運行,這種訓練需要數(shù)周時間。
 AlphaFold解決蛋白質(zhì)折疊問題的方法
AlphaFold解決蛋白質(zhì)折疊問題的方法DeepMind 在2018年首次使用最初版本的 AlphaFold 參加 CASP13,在參賽者中獲得了最高的準確度,隨后又在《自然》雜志上發(fā)表了一篇關(guān)于 CASP13 方法及相關(guān)代碼的論文,這篇論文繼續(xù)啟發(fā)了其他工作和社區(qū)開發(fā)的開源實現(xiàn)。
現(xiàn)在,DeepMind開發(fā)的新的深度學習架構(gòu)已經(jīng)推動了 CASP14方法的變化,使之能夠達到前所未有的精確度。這些方法的靈感來自生物學、物理學和機器學習領(lǐng)域,當然還有過去半個世紀許多蛋白質(zhì)折疊領(lǐng)域的科學家的工作。
一個折疊的蛋白質(zhì)可以被認為是一個“空間圖形”,其中殘基是節(jié)點和邊連接的。

這張圖對于理解蛋白質(zhì)內(nèi)部的物理相互作用以及它們的進化歷史是很重要的。
對于在 CASP14上使用的最新版本的 AlphaFold,研究人員創(chuàng)建了一個基于注意力的神經(jīng)網(wǎng)絡(luò)系統(tǒng),通過端到端的訓練來解釋這個圖的結(jié)構(gòu),同時推理出它正在構(gòu)建的隱式圖。它通過使用多重序列對齊 (MSA) 和氨基酸殘基對的表示來精化這個圖結(jié)構(gòu)。
通過迭代這個過程,系統(tǒng)可以對蛋白質(zhì)的基本物理結(jié)構(gòu)進行準確的預測,并能夠在幾天的時間內(nèi)確定高度精確的結(jié)構(gòu)。
此外,AlphaFold 還可以使用內(nèi)部置信度來預測每個預測的蛋白質(zhì)結(jié)構(gòu)的哪些部分是可靠的。
這個系統(tǒng)所使用的數(shù)據(jù)包括來自蛋白質(zhì)數(shù)據(jù)庫的大約170,000個蛋白質(zhì)結(jié)構(gòu),以及包含未知結(jié)構(gòu)的蛋白質(zhì)序列的大型數(shù)據(jù)庫。它使用了大約128個 TPU v3 (大致相當于100-200個 GPU) ,只訓練了幾周時間,在當今機器學習領(lǐng)域中使用的大多數(shù)SOTA模型中來說是一個相對較少的計算量。
CASP:蛋白質(zhì)奧林匹克競賽
1994年,John Moult 教授和 Krzysztof Fidelis 教授創(chuàng)立了 CASP (Critical Assessment of Structure Prediction) 作為兩年一次的盲選評估,以促進研究并建立蛋白質(zhì)結(jié)構(gòu)預測的最新水平。
CASP 是評估預測技術(shù)的標準。更重要的是,CASP 選擇那些最近才經(jīng)過實驗確定的蛋白質(zhì)結(jié)構(gòu)(有些在評估時仍在等待確定)作為研究小組測試其結(jié)構(gòu)預測方法的目標; 而這些結(jié)構(gòu)的預測方法并沒有提前公布。參與者必須直接預測蛋白質(zhì)的結(jié)構(gòu),這些預測隨后會在可用時與實驗數(shù)據(jù)進行比較。

CASP 用來測量預測準確性的主要指標是 GDT(Global Distance Test ) ,其范圍是從0-100。簡單地說,GDT 可以大致地被認為是氨基酸殘基在閾值距離內(nèi)與正確位置的百分比。根據(jù) Moult 教授的說法,90分左右的 GDT 可以被認為是與實驗方法得到的結(jié)果相競爭的。

在今天公布的第14屆 CASP 評估結(jié)果中,最新的 AlphaFold 系統(tǒng)在所有目標中總體得分中位數(shù)為92.4 GDT。這意味著預測平均誤差(RMSD)約為1.6埃(1埃等于0.1納米),相當于一個原子的寬度(或0.1納米),即使對于最難的蛋白質(zhì)目標,即那些最具挑戰(zhàn)性的自由建模分類,AlphaFold 也可以達到87.0 GDT 的中位數(shù)。

在接受檢驗的近100個蛋白靶點中,AlphaFold 對三分之二的蛋白靶點給出的預測結(jié)構(gòu)與實驗手段獲得的結(jié)構(gòu)相差無幾。CASP 創(chuàng)始人 Moult 教授表示,在有些情況下,已經(jīng)無法區(qū)分兩者之間的區(qū)別是由于AlphaFold的預測出現(xiàn)錯誤,還是實驗手段產(chǎn)生的假象。

這些令人興奮的結(jié)果為生物學家打開了使用深度學習計算結(jié)構(gòu)預測作為科學研究的核心工具的潛力,DeepMind 的方法可能特別有助于預測重要類別的蛋白質(zhì)結(jié)構(gòu),如膜蛋白等。
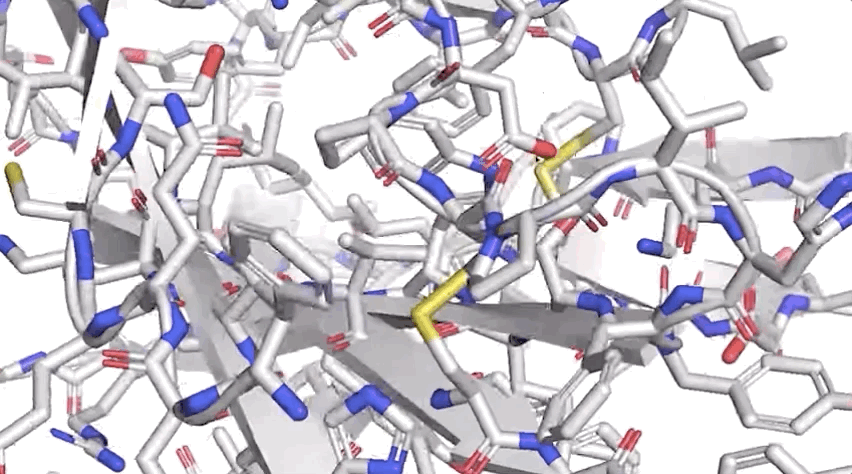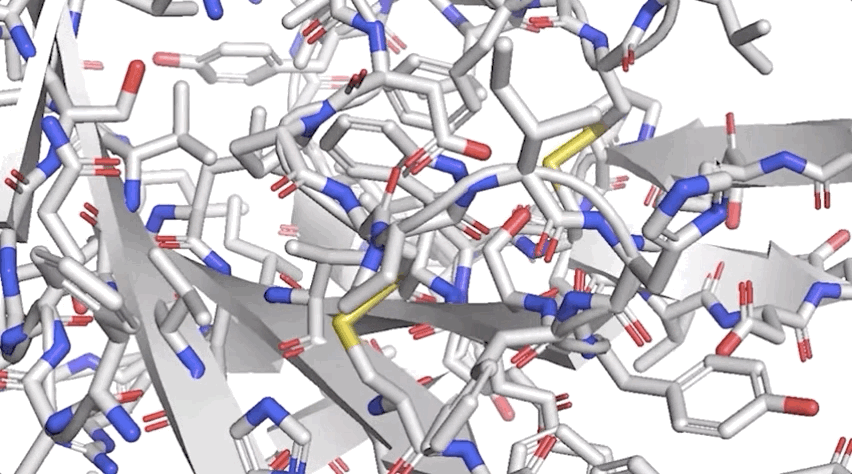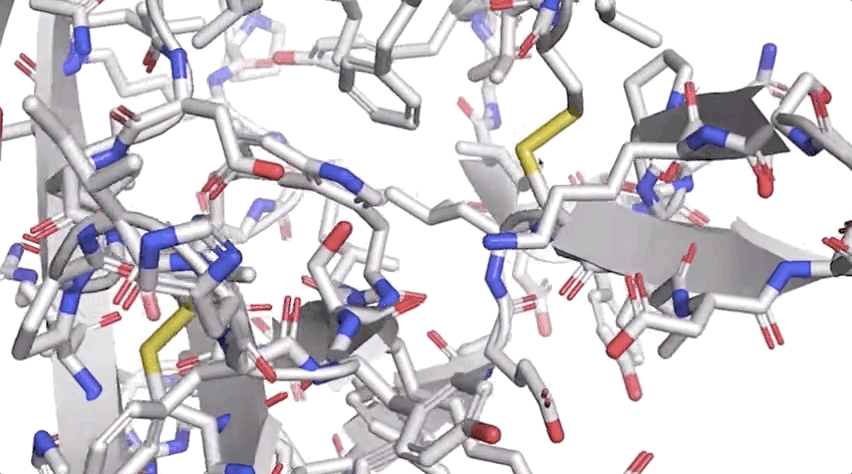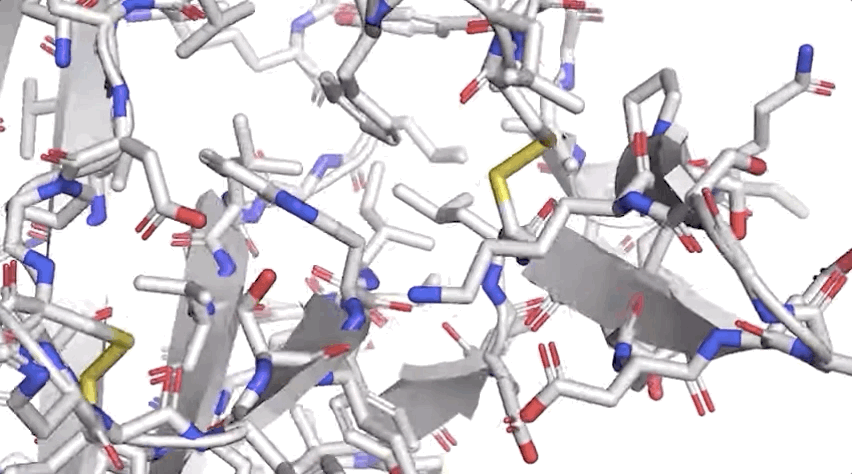
 圖:ALPHAFOLD 預測了與實驗結(jié)果相對應(yīng)的高度精確的結(jié)構(gòu)
圖:ALPHAFOLD 預測了與實驗結(jié)果相對應(yīng)的高度精確的結(jié)構(gòu)歷史性突破!AlphaFold將“改變一切”
如果把基因組序列比喻為標識一個人的身份信息,蛋白質(zhì)的三維結(jié)構(gòu)就是一個人的身形容貌。
預測蛋白質(zhì)結(jié)構(gòu)變化的重要意義在于,包括癌癥、癡呆等幾乎所有疾病,都與細胞內(nèi)蛋白質(zhì)結(jié)構(gòu)變化相關(guān),如果能夠掌握蛋白質(zhì)結(jié)構(gòu)的變化,將對疾病的預防、治療等帶來重要影響。
通常情況下,識別單個蛋白質(zhì)的結(jié)構(gòu)需要耗費科學家數(shù)年時間。如今,AlphaFold能在幾天內(nèi)提供精確到一個原子的結(jié)果。

此舉將極大地加速人類對細胞組成部分的理解,對包括新冠肺炎在內(nèi)所有疾病的研究均有所幫助。
在冠狀病毒的結(jié)構(gòu)中,像皇冠一樣的刺突稱為刺突糖蛋白(Spike Glycoprotein,以下簡稱 S 蛋白),是結(jié)合人體細胞上相應(yīng)受體的罪魁禍首;E 蛋白是包膜蛋白,將病毒內(nèi)部的遺傳物質(zhì)包裹起來;還有膜蛋白(M 蛋白)和核衣殼蛋白(N 蛋白)等結(jié)構(gòu)。
 冠狀病毒結(jié)構(gòu)示意圖
冠狀病毒結(jié)構(gòu)示意圖三維結(jié)構(gòu)的解析對于新冠病毒致病機理和藥物設(shè)計具有非常重要的先決意義。
基于AlphaFold的新突破,人類未來也可能更快地發(fā)現(xiàn)更先進的新藥物。
CASP聯(lián)合創(chuàng)始人、馬里蘭大學帕克分校計算生物學家John Moult直言,“這是一件大事,在某種程度上來說,(蛋白質(zhì)折疊)問題解決了。”
挑戰(zhàn)賽評委之一的進化生物學家Andrei Lupas進一步表示,“這將改變醫(yī)學,這將改變研究,這將改變生物工程,這將改變所有一切。”
知名領(lǐng)域?qū)<襇ohammed AlQuraishi發(fā)推稱;“它們令人震驚——deepmind似乎已經(jīng)解決了蛋白質(zhì)結(jié)構(gòu)預測問題。”

谷歌CEOSundar Pichai在當天也在推特上分享了這一消息并表示:
“DeepMind難以置信的用AI進行蛋白質(zhì)折疊預測的突破,將幫助我們更好地理解生命的最根本的根基,并幫助研究人員應(yīng)對新的和更難的難題,包括應(yīng)對疾病和環(huán)境可持續(xù)發(fā)展。”

參考鏈接:
https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology

(聲明:本文僅代表作者觀點,不代表新浪網(wǎng)立場。)

作者簡介

新智元
作者文章
Arm中國“奪帥”羅生門繼續(xù)!吳雄昂稱Arm罷免決議無效

盡管已經(jīng)賣身英偉達了,但Arm公司和吳雄昂的矛盾仍未解決。在最新的采訪中,吳雄昂表示:Arm及厚樸投資等無權(quán)罷免他作為Arm中國的首席執(zhí)行官一職。吳雄昂還透露,成立Alphatecture基金,是‘得到董事會支持的’。
推薦閱讀
- 被阿里耽誤的蝦米的一生
-

- 真正的蝦米音樂粉絲,只要打開自己的App界面就可以證明。注冊年限、聽過的歌曲數(shù)量,無一不是最好的證明。詳細>>
- 游戲主業(yè)增長低于預期,金山軟件遭遇估值挑戰(zhàn)
-

- 對于投資者來說,看好云賽道就去投資金山云,看好線上辦公就去投資金山辦公,而看好游戲就未必會投資金山軟件了。詳細>>
- 在線教育融資2020:資本青睞的獨角獸外,僅剩沉默大多數(shù)
-

- 當教學剛需遇到線下場景暫時冰封,今年上半年,在線教育迎來前所未有的流量大潮,下沉市場對在線教育的認知度和認可度也得到明顯提升。詳細>>
- 特斯拉摸著石頭過河 汽車老前輩們摸著特斯拉過河
-

- 詳細>>