




近日,德爾塔毒株在南京、鄭州等地引發新一輪疫情。中國疾控中心研究員馮子健指出,德爾塔有潛伏期和傳代間隔縮短的特點,如果沒有疫苗,可能病人的數量大約 3 天就會增加到五到七倍。在美國,該毒株導致確診病例出現新一輪的增長,來自美國疾病預防和控制中心(CDC)的一份文件稱,德爾塔毒株傳染能力比季節性流感更強,與水痘相當。
德爾塔毒株為什么如此兇險?研究人員正在努力剖析新冠病毒的生命周期,看看它有哪些逃逸免疫系統的招數。
本文轉載自公眾號“Nature Portfolio”
原文作者:Megan Scudellari
新冠病毒(SARS-CoV-2)有著很不一般的“糖衣”。“太驚人了。”Rommie Amaro 盯著她為新冠病毒標志性刺突蛋白做的計算機模擬說道。這些刺突蛋白從新冠病毒的表面突起,包裹在聚糖(glycan)這種糖分子里。
“如果它包裹在這些聚糖中,那幾乎就無法識別。”加州大學圣迭戈分校的計算生物物理化學家 Amaro 說。
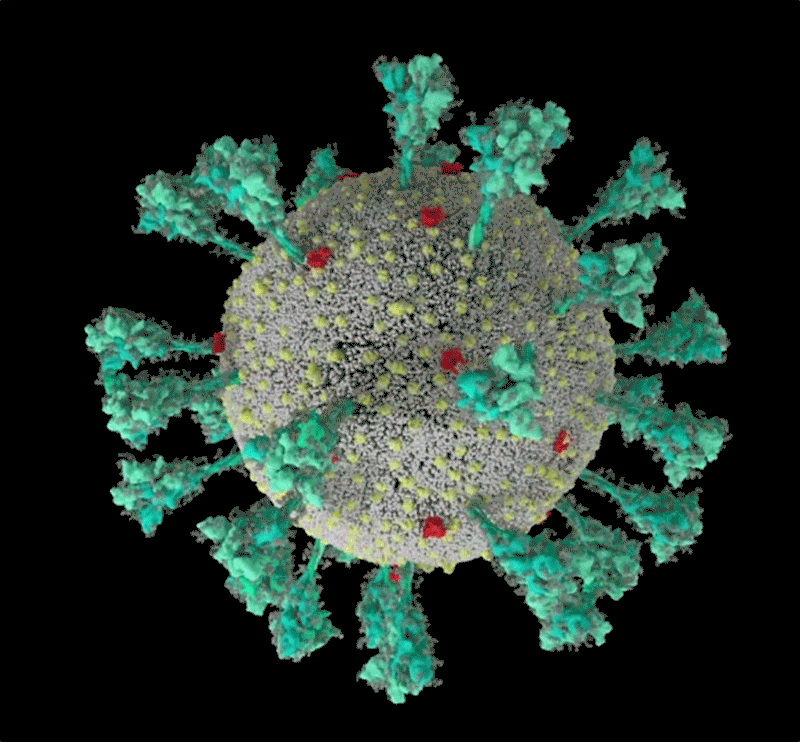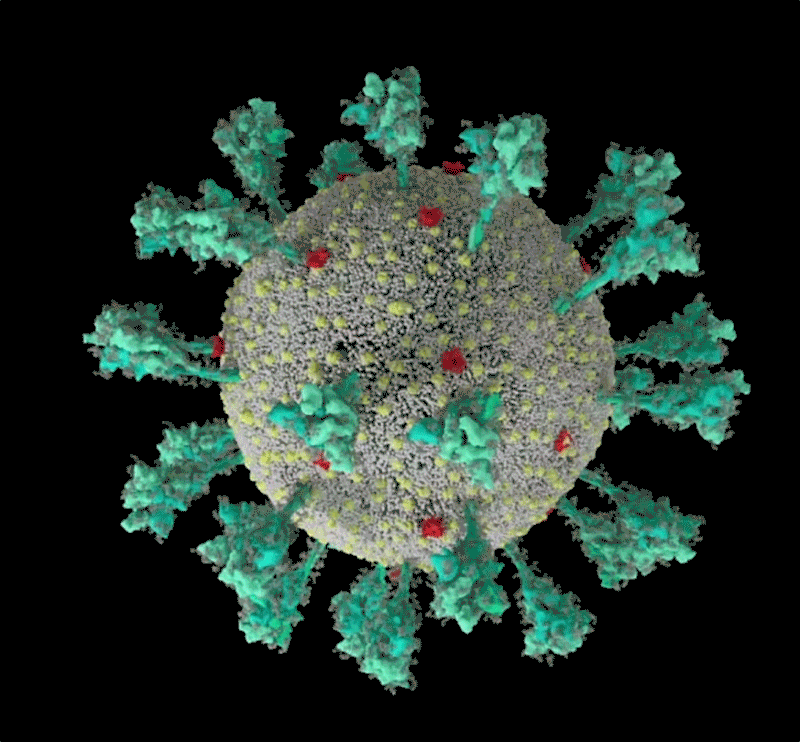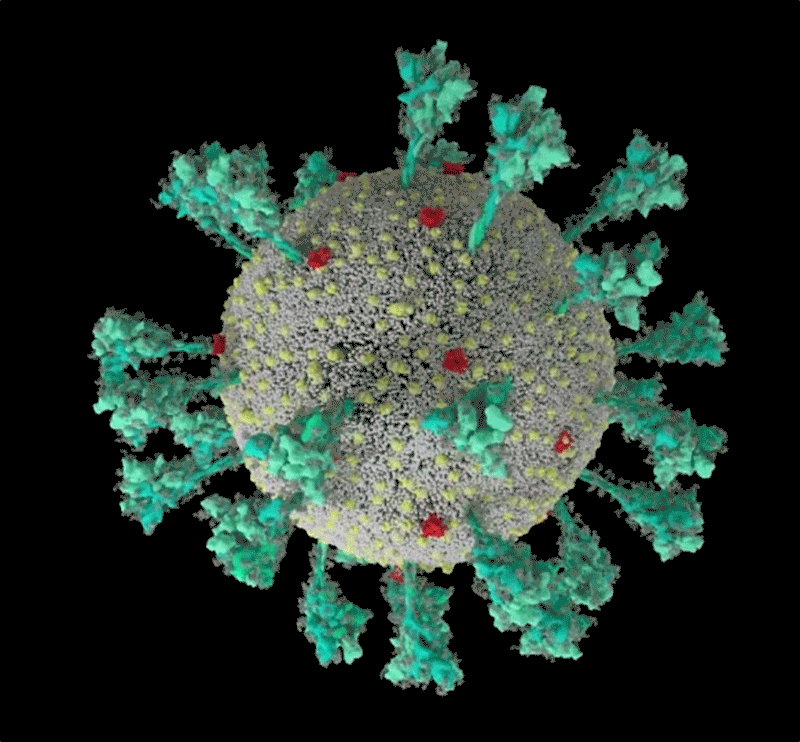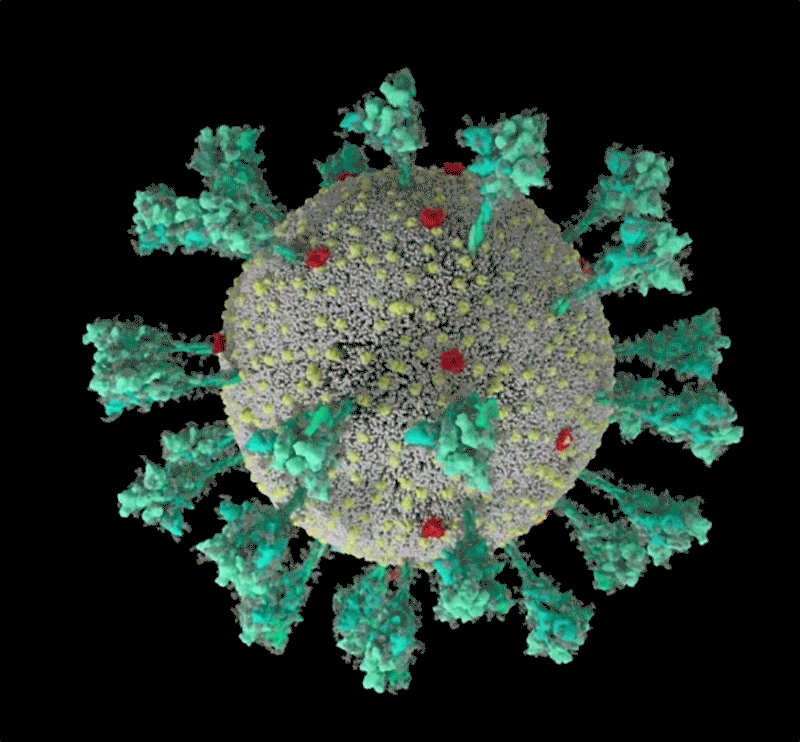
新冠病毒(SARS-CoV-2)結構的計算機模擬。來源:Janet Iwasa, University of Utah
為了偽裝,許多病毒都會用聚糖包裹表面蛋白,就像披著羊皮的狼,逃過人體免疫系統的監視。去年,Amaro 的實驗小組與合作者基于結構和遺傳學數據,利用超級計算機進行逐原子渲染,對這層“羊皮”進行了迄今精度最高的可視化。2020 年 3 月 22 日,她在推特上發布了模擬結果。還不到一個小時,一位研究人員在評論中問道,從刺突蛋白頂部突出的無包裹裸露環狀結構是什么?
Amaro 被問倒了。但 10 分鐘后,得克薩斯大學奧斯汀分校的結構生物數學家 Jason McLellan 回復道,這個裸露的環狀結構是一個受體結合結構域(RBD),是刺突蛋白與人細胞表面受體結合的三個區域之一(見“藏起來的刺突蛋白”)。

來源:Structural image from Lorenzo Casalino, Univ。 California, San Diego (Ref。 1); Graphic: Nik Spencer/Nature
在 Amaro 的模擬中,當 RBD 上升到這團聚糖的頂部時,兩個聚糖會突然將其鎖住,就像自行車的撐腳架一樣。如果 Amaro 改變計算機模型中的聚糖,RBD 就會倒塌。McLellan 的團隊設法在實驗室進行了同樣的實驗。到 2020 年 6 月,這些合作者已經報道稱,改變這兩個聚糖會降低刺突蛋白與人細胞受體的結合力[1]——之前從未在冠狀病毒中發現過這種作用,McLellan 說。剪掉這兩個聚糖或能降低新冠病毒的感染性,Amaro 說,可惜研究人員尚未找到這么做的方法。
自新冠疫情出現以來,研究人員已經對該病毒如何感染細胞有了詳盡的認識。通過解析整個感染過程,研究人員希望憑借改良的療法或疫苗抑制感染,同時搞清楚 Delta 等最新毒株的傳播力為何會上升。
過去19個月的不懈努力,加之數十年的冠狀病毒研究,我們正逐漸解開新冠病毒是如何一步步侵入人體細胞的(見“新冠病毒的生命周期”)。研究人員確定了新冠病毒能以出人意料的力量抓住人體細胞,而后立即遁形的關鍵適應機制。隨后,新冠病毒離開細胞時又會使出關鍵一招,令其病毒顆粒能繼續感染更多人體細胞。這是新冠病毒用來迅速傳播、奪人性命的一些招數。帝國理工學院病毒學家 Wendy Barclay 說:“這是新冠病毒難以遏制的原因。”
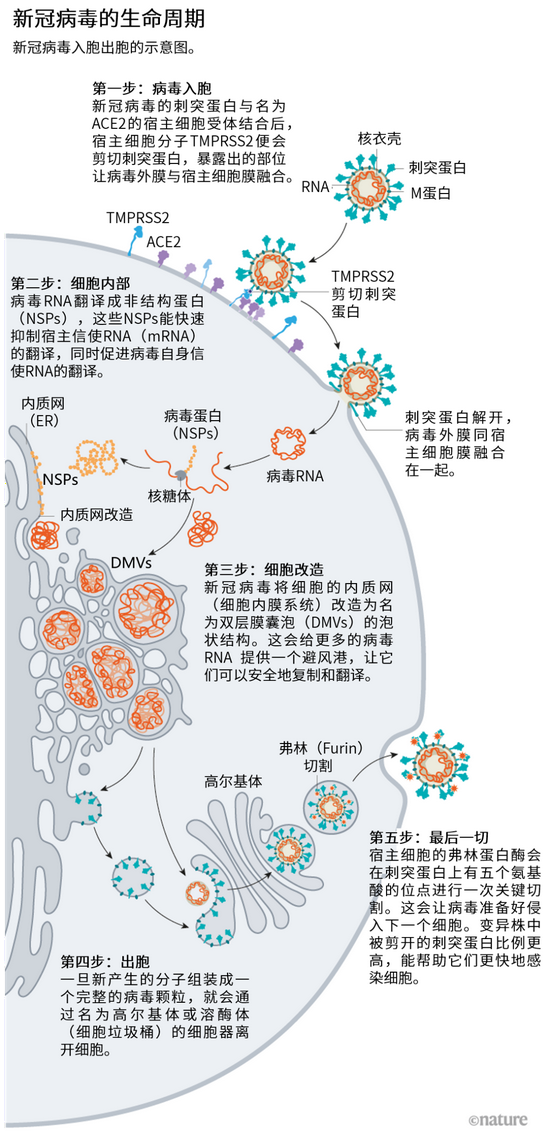
蓄“刺”待發
一切從刺突蛋白開始。每個新冠病毒顆粒(virion)的表面有 24-40 個任意排列的刺突蛋白,這些刺突蛋白是病毒與人細胞融合的關鍵[2]。與流感病毒等其他病毒表面的剛性融合蛋白相比,新冠病毒的刺突蛋白非常靈活,能在三個點像鉸鏈一樣活動,德國馬克斯·普朗克生物物理學研究所生物化學家 Martin Beck 和同事在 2020 年 8 月發表的一項研究中指出[3]。
如此一來,刺突蛋白就能任意掉頭、搖擺、轉動,這有利于它對細胞表面進行掃視,或是用多個刺突蛋白與一個人體細胞相結合。目前其他冠狀病毒尚無類似實驗數據,但由于刺突蛋白序列在演化上高度保守,可以推測所有冠狀病毒都具有這一特點,Beck 說。
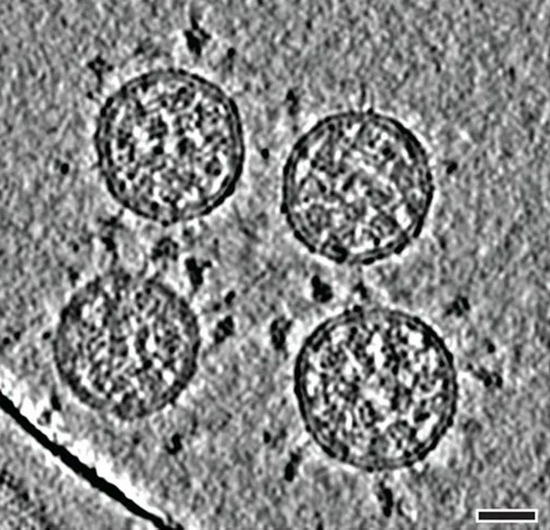
新冠病毒顆粒的冷凍電鏡斷層掃描成像。(比例尺 30 納米。)來源:B。 Turoňová et al。/Science
研究人員在疫情早期就證實了新冠病毒刺突蛋白的 RBD 能與 ACE2 受體結合。ACE2 受體是一種常見的蛋白,廣泛分布于人類喉部和肺部的大部分細胞表面。這個受體還是導致嚴重急性呼吸系統綜合征的 SARS 病毒(SARS-CoV)的入胞點。但是,新冠病毒與 ACE2 的親和力更好,估計是 SARS 病毒的 2-4 倍[4],這是因為 RBD 的多種變化能穩定其與病毒結合的熱點位置[5]。
危險的新冠病毒變異株容易在刺突蛋白的 S1 亞基上攜帶突變,S1 亞基包含 RBD,并負責病毒與 ACE2 受體的結合(刺突蛋白的 S2 亞基則能促進病毒外膜與宿主細胞膜的融合。)
以 Alpha 變異株為例,它在刺突蛋白序列上有 10 個突變,導致 RBD 更有可能保持“向上”的狀態[6]。“這能讓病毒更易進入細胞。”美國北卡羅來納州杜克人類疫苗研究所的結構生物學家 Priyamvada Acharya 說。Acharya 正在研究刺突蛋白的突變。
目前 Delta 變異株正在世界各地傳播,它在 S1 亞基上有多個突變,包括 RBD 上的 3 個突變,這 3 個突變似乎不僅能提高 RBD 與 ACE2 的親和力,還能提高其逃逸免疫系統的能力[7]。
限定入口
病毒刺突蛋白與 ACE2 結合后,宿主細胞表面的其他蛋白就會啟動病毒外膜與細胞膜融合的過程(見“病毒入胞近景”)。
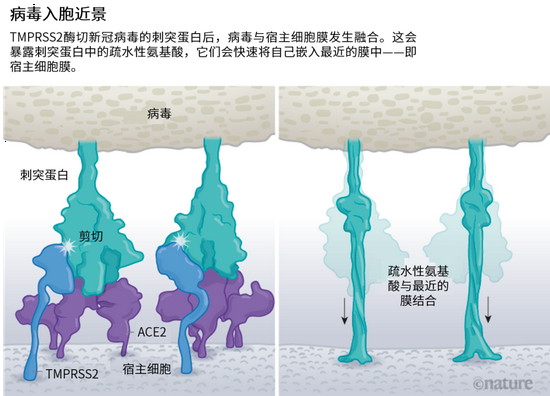
SARS 病毒會利用兩個宿主蛋白酶中的一個入胞:TMPRSS2 (讀作 tempress two)或組織蛋白酶 L。TMPRSS2 的入胞途徑更快,但 SARS 病毒經常通過核內體侵入,核內體是一種脂質包裹的囊泡,這個途徑依賴組織蛋白酶 L。不過,如果病毒顆粒從這一途徑進入,就會被抗病毒蛋白逮個正著。
新冠病毒之所以不同于 SARS 病毒,就在于它能更快地利用 TMPRSS2。TMPRSS2 是呼吸道細胞表面大量存在的一種酶。TMPRSS2 會先酶切刺突蛋白 S2 亞基上的一個位點[8]。這個剪切點會暴露一串疏水性氨基酸,暴露后的疏水性氨基酸會迅速嵌入最近的膜中——即宿主細胞膜。隨后,展開的刺突蛋白會折疊起來,像拉鏈一樣,迫使病毒外膜與細胞膜融合。
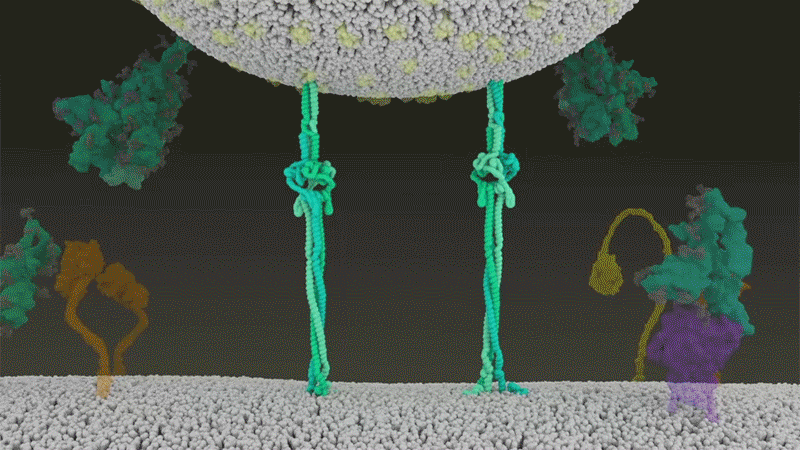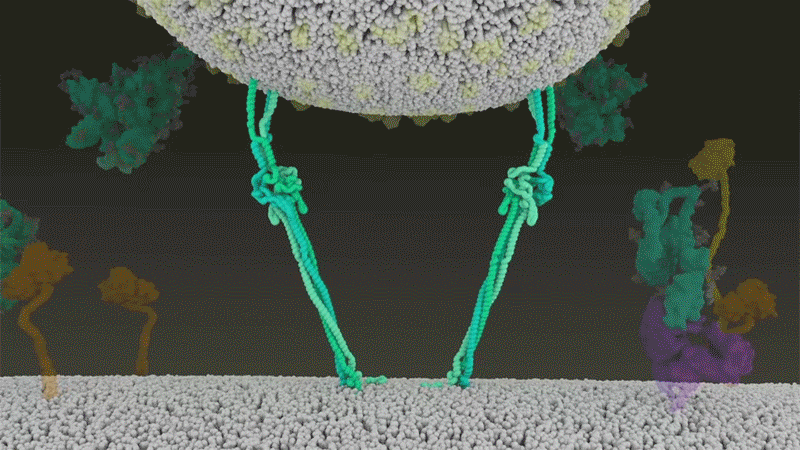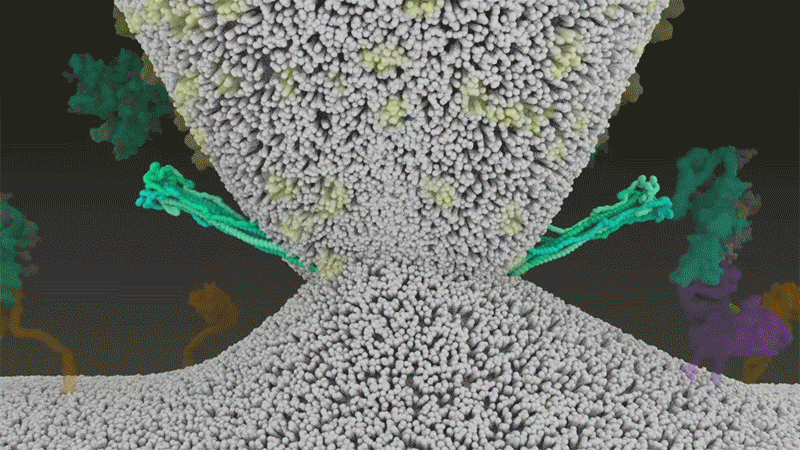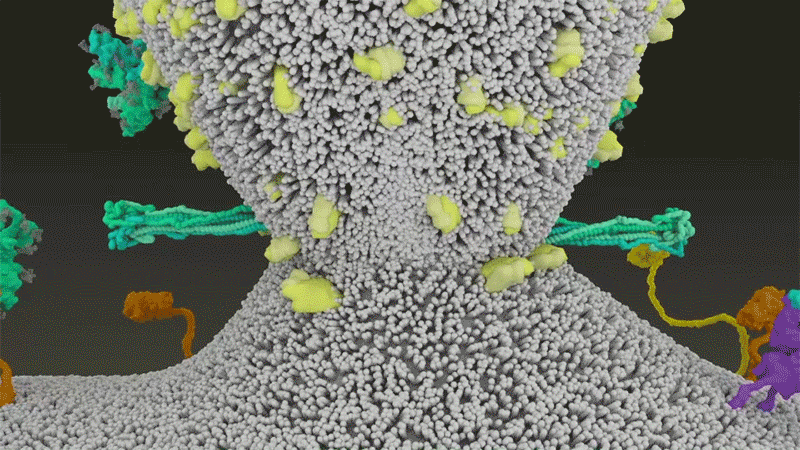
新冠病毒與細胞融合過程的動畫展示。來源:Janet Iwasa, University of Utah
隨后,新冠病毒將基因組直接注射到宿主細胞內。通過采取這種彈簧式的侵入方式,新冠病毒比 SARS 病毒的感染更快,而且不會被核內體逮住,Barclay 和她在英國帝國理工學院的同事在 4 月發表的一項研究中描述道[9]。
新冠病毒能利用 TMPRSS2 實現快速入胞,解釋了為何瘧疾藥物氯喹一開始在實驗室研究中表現良好,但在治療 COVID-19 的臨床試驗中無效[10]。原來氯喹利用的細胞完全依賴組織蛋白酶實現核內體侵入。“新冠病毒在人類氣道中傳播和復制時不會用到核內體,所以氯喹這種核內體干擾藥物在真實人體中效果就不大了。”Barclay 說。
這一發現還指出,蛋白酶抑制劑是一種很有希望的治療選項,可以防止病毒利用 TMPRSS2、組織蛋白酶 L 或其他蛋白酶進入宿主細胞。camostat mesylate 是一種 TMPRSS2 抑制劑,日本已將其批準用于治療胰腺炎。這種抑制劑能阻斷病毒進入肺部細胞[8],但無法在初步臨床試驗中提高病人的治療轉歸[11]。
“依我看,我們應當將這種蛋白酶抑制劑作為廣譜抗病毒藥物,防止新的疾病發展為大流行,將其遏制在萌芽階段。”德國靈長類研究中心感染生物學部主任 Stefan P?hlmann 說。P?hlmann 領導開展了 ACE2 結合和 TMPRSS2 途徑的研究。
致命競爭
接下來的感染步驟就沒那么清晰了。“進入細胞之后就有很多黑箱。不確定性和各種假說也更多了。”美國猶他大學的化學家 Janet Iwasa 說。Iwasa 正在制作一個解釋新冠病毒生命周期的帶注釋的動畫。
就在新冠病毒將 RNA 基因組注射到宿主細胞后,細胞質核糖體會將兩個病毒 RNA 片段翻譯成氨基酸長鏈,這些氨基酸長鏈再被切割出 16 個蛋白質,包括許多參與 RNA 合成的蛋白質。隨后會產生更多的 RNA,這些 RNA 編碼 26 個已知的病毒蛋白,包括用來制造新病毒顆粒的結構蛋白(比如刺突蛋白)和其他協助蛋白。這樣,病毒就能大量產生其自身的信使 RNA(mRNA)拷貝,但它還需要細胞機器來將這些 mRNA 翻譯成蛋白質。
新冠病毒有很多將細胞機器占為己用的策略。病毒學家 Noam Stern-Ginossar 和她在以色列魏茨曼科學研究所的團隊重點研究了新冠病毒抑制宿主 mRNA 翻譯、促進自身 mRNA 翻譯的三種機制。雖然這三種機制并非新冠病毒所獨有,但這些作用的結合、速度和程度看來確實是獨一無二的,Stern-Ginossar 說。
第一,新冠病毒會清除競爭對手:病毒蛋白 Nsp1 是新冠病毒抵達時首先被翻譯的蛋白之一,它會把宿主蛋白質招募起來,系統性地切割所有不帶病毒標記的細胞 mRNA。如果 Stern-Ginossar 的團隊將同樣的病毒標記放在宿主 mRNA 的末端,這個 mRNA 就不會被切割[12]。
第二,感染會讓細胞內全部蛋白質翻譯減少 70%。Nsp1 依然是搗蛋分子,這一次它能阻斷核糖體的入胞渠道,讓 mRNA 無法進入,兩個研究團隊分別得出了以上結論[13,14]。剩下不多的翻譯能力全被用來翻譯病毒 RNA,Stern-Ginossar 說。
第三,新冠病毒會關閉細胞的預警系統。這有很多方式,但 Stern-Ginossar 的團隊發現了新冠病毒的一個明確機制:病毒讓細胞 mRNA 無法離開細胞核,包括提醒免疫系統注意感染的蛋白質的指令。另一個團隊證實了這個結果,再次將矛頭指向 Nsp1:這個蛋白似乎會阻塞離開細胞核的通道,一個不讓逃走[15]。
由于基因轉錄本無法離開細胞核,因此受感染的細胞不會釋放許多干擾素——干擾素是提醒免疫系統注意病毒的信號蛋白。新冠病毒關閉這一預警系統的速度尤其快:和其他呼吸道病毒相比,包括 SARS 病毒和呼吸道合胞病毒,新冠病毒感染后誘導的干擾素水平顯著降低[16]。今年 6 月,研究人員報道了 Alpha 變異株的突變似乎能更有快地減少干擾素的產生[17]。
“新冠病毒明顯是個動作很快的病毒,具有非常獨特的能力,能從源頭上擾亂免疫系統識別病毒和抵抗感染的能力。”Stern-Ginossar 說。等到免疫系統真正發現有病毒時,病毒數量已經太多了,致使免疫應答蛋白有時會比平常更快地充斥在血液中,而這可能對人體有害。臨床醫生在疫情很早就發現,有些發展成重癥的 COVID-19 患者除了受到病毒本身的襲擊外,過度激活的免疫應答也造成了一定傷害。一些經證明有效的療法專門抑制這種免疫應答。
最強改造王
新冠病毒接管宿主細胞翻譯后,它就開始喧賓奪主了,朝著有利自己的方向大肆改造細胞內部和表面。
首先,一些新制造的刺突蛋白會抵達宿主細胞表面,突破宿主細胞膜。它們還會在那里激活一條宿主鈣離子通道,在細胞表面分泌一層脂肪膜——肌肉細胞等自然融合的細胞上也會發現這種膜。這時,受感染細胞與表達 ACE2 的相鄰細胞融合,發展為最多包含 20 個細胞核的單個大呼吸道細胞。

表達新冠病毒刺突蛋白(綠色)的細胞內可見融合細胞結構(合胞體)。藍色為細胞核,紅色為細胞骨架。來源:Mauro Giacca
這些融合結構被稱為合胞體(syncytia),HIV 和單純皰疹病毒等病毒感染會誘導形成合胞體,但 SARS 病毒就不會,倫敦國王學院的分子生物學家 Mauro Giacca 說。Giacca 領導的團隊在 4 月發表了這項研究結果[18]。他的假設是,形成合胞體能讓受感染細胞存活更長時間,制造更多病毒顆粒。“這不是一個會肇事逃逸的病毒。”他說。“它會一直存在。”中國醫學科學院研究員孫強領導的另一支團隊發現,新冠病毒感染的一些細胞甚至會與淋巴細胞形成合胞體,淋巴細胞是人體自身的免疫細胞[19]。這其實是腫瘤細胞的免疫逃逸機制,而非病毒的機制,提示我們受感染細胞會抓住周圍的免疫細胞并與之融合,輕而易舉地逃過免疫系統的偵查。
細胞內部的變化就更多了。內質網(ER)是參與蛋白合成與轉運的平面膜系統,和其他冠狀病毒一樣,新冠病毒會將又長又細的內質網變成一個具有雙層膜的球體,就好像內質網在吹泡泡一樣。這些雙層膜囊泡(DMVs)或許能為病毒 RNA 提供一個復制和翻譯的避風港,保護其不被細胞內的天然免疫傳感器發現。當然,這個假說仍有待驗證。
參與制造 DMVs 的蛋白或許是很好的藥物靶點,因為它們對病毒復制好像起著不可或缺的作用。比如,TMEM41B 這種宿主蛋白必須被用來調動膽固醇和其他脂質來擴大內質網膜,以便所有病毒成分都能進入其中[20]。“把 TMEM41B 拿走會對感染產生很大影響。”參與這項研究的得克薩斯大學醫學部的新冠病毒研究員 Vineet Menachery 說。此外,新冠病毒跨膜蛋白 Nsp3 也可作為藥物靶點:它會在 DMVs 上形成冠狀孔,將制造好的病毒 RNA 轉運出去[21]。
大部分具有外層膜(即包膜)的病毒獲得這一特征的方式是直接在細胞邊緣組裝,并在離開的路上選擇一些細胞自己的質膜。但新制造好的冠狀病毒蛋白會選擇另一個途徑。
多年來的證據已經表明,冠狀病毒會通過高爾基復合體轉出細胞。高爾基復合體是具有郵局功能的細胞器,能將分子包在膜內,運送至細胞其他部位。病毒會在那里從高爾基體膜形成一個脂質包膜;新形成的病毒顆粒再在高爾基囊泡中轉運到細胞表面,并分泌到胞外,美國約翰斯·霍普金斯大學病毒學家、生物學家 Carolyn Machamer 說。Machamer 研究冠狀病毒已經有 30 年了。
但在去年 12 月,美國國家心臟、肺和血液研究所的細胞生物學家 Nihal Altan-Bonnet 和她的同事報道,他們發現了冠狀病毒會通過溶酶體離開細胞——溶酶體是細胞的垃圾桶,擁有很多能分解細胞成分的酶[22]。阻斷基于高爾基體的分泌途徑似乎不會影響釋放的感染病毒數量,Altan-Bonnet 說。她團隊的證據[22]顯示,病毒蛋白在內質網出芽形成包膜,再占用溶酶體離開細胞。目前,該團隊正在檢測阻斷溶酶體出胞過程的抑制劑是否能成為潛在的抗病毒藥物。
無論是通過高爾基體還是溶酶體離開細胞,都比通過質膜出芽要慢、效率也更低,所以研究人員不明白新冠病毒為什么會這么做。Machamer 推測,相比質膜,來自高爾基體或溶酶體的包膜的脂質組成好像對新冠病毒更有利。“如果我們能更好地理解這個部分,就有很大的機會找到新的抗病毒療法。”她說。
最后一切
在離開細胞時,還有一步讓這個病毒成為傳染王:在有 5 個氨基酸的位點進行快速切割能讓該病毒準備好攻擊下一個目標。
其他冠狀病毒在刺突蛋白 S1 和 S2 亞基的連接處只有一個精氨酸,新冠病毒卻有 5 個連著的氨基酸:脯氨酸、精氨酸、精氨酸、丙氨酸、精氨酸。“由于這個位點非常特殊,我們就盯著它,最后發現這個位點確實是侵入肺部細胞的關鍵。”P?hlmann 說。2020 年 5 月,他和同事報道了名為弗林(furin)的宿主細胞蛋白能識別并切割這個氨基酸鏈,而且這種切割對于新冠病毒快速進入人肺部細胞是“至關重要的”[23]。
這不是研究人員第一次在病毒上發現弗林切割位點;致病性很高的禽流感病毒也有這個位點,Barclay 說。當一位同事把培養的自然失去弗林切割位點的新冠病毒變異株給到 Barclay 時,她的團隊發現感染該毒株的雪貂比感染大流行毒株的雪貂脫落的病毒顆粒更少,而且不會將病毒傳給周圍的動物[9]。正當 Barclay 的團隊準備在 2020 年 9 月的預印本論文中報道該結果時,荷蘭的一項研究也發現,擁有完整弗林切割位點的冠狀病毒進入人氣道細胞的速度比沒有弗林切割位點的更快[24]。
研究人員推測弗林會在病毒顆粒組裝過程中或是釋放前切割該位點。這個時間點解釋了新冠病毒為何會通過高爾基體或溶酶體離開細胞,芝加哥洛約拉大學的病毒學家 Tom Gallagher 說。“這個病毒一經組裝就會移動到周圍都是弗林蛋白酶的細胞器中。”
通過剪切 S1 和 S2 亞基之間的連接鍵,弗林酶切讓病毒顆粒的刺突蛋白松開,以便它們在進入細胞時對 TMPRSS2 的二次切割產生反應,這次切割暴露出的疏水性區域會快速將自己嵌入宿主細胞膜內,Gallagher 說。如果刺突蛋白沒有被弗林蛋白預先切開——有時也不會被切開——它們就會繞開 TMPRSS2,通過更慢的核內體途徑進入細胞,或是根本不進入細胞。
Alpha 和 Delta 變異株的弗林切割位點都發生了變化。Alpha 變異株將本來的脯氨酸替換成組氨酸(P681H);Delta 變異株則替換成了精氨酸(P681R)。這兩個變化都會減少序列的酸性;而且氨基酸鏈堿性越強,它們被弗林識別切割的效果也更好,Barclay 說。“我們的假設是,這體現出新冠病毒的傳遞能力增強了。”
更多弗林酶切意味著更多刺突蛋白準備好進入人體細胞。SARS病毒只有不到 10% 的刺突蛋白做好了這種準備,Menachery 說。Menachery 的實驗小組一直在量化這些做好準備的刺突蛋白,但研究成果尚未發表。對新冠病毒來說,這個比例上升至 50%,而 Alpha 毒株超過了 50%,該團隊發現,在傳染性很強的 Delta 毒株中,75% 以上的刺突蛋白準備好繼續感染人體細胞。
走向未知
科研界其實才剛剛開始理解新冠病毒。一些關鍵的未知數還包括:與每個刺突蛋白結合所需的 ACE2 受體數量;S2 位點究竟是何時被 TMPRSS2 切割的;病毒外膜與細胞膜融合所需的刺突蛋白數量,McLellan 說,而這些還只是入胞的問題。2020 年 4 月,加州大學舊金山分校的一個團隊鑒定出了新冠病毒與人類蛋白質相互作用的至少 332 種方式[25]。
想要趕上這個快速變異的病毒很不容易。但專家一致認為,迄今發現的主要是變異與病毒傳播速度的相關性,而不是與病毒對宿主傷害的相關性。7 月的一項研究報道,Delta 變異株在肺部和喉部的生長速度比之前的變異株快很多[26]。
但是,現在還不確定 Delta 攜帶的突變如何以這種方式為其毒力加碼,Stern-Ginossar 說,“許多實驗室正在竭力回答這些問題。”
參考文獻:
1。 Casalino, L。 et al。 ACS Cent。 Sci。 6, 1722–1734 (2020)。
2。 Ke, Z。 et al。 Nature 588, 498–502 (2020)。
3。 Turoňová, B。 et al。 Science370, 203–208 (2020)。
4。 Nguyen, H。 L。 et al。 J。 Phys。 Chem。 B124, 7336–7347 (2020)。
5。 Shang, J。 et al。 Nature581, 221–224 (2020)。
6。 Gobeil, S。 M。-C。 et al。 Science https://doi.org/10.1126/science.abi6226 (2021)。
7。 Khateeb, J。, Li, Y。 & Zhang, H。 Crit。 Care25, 244 (2021)。
8。 Hoffmann, M。 et al。 Cell 181, 271–280 (2020)。
9。 Peacock, T。 P。 et al。 Nature Microbiol。 6, 899–909 (2021)。
10。 Wang, M。 et al。 Cell Res。30, 269–271 (2020)。
11。 Gunst, J。 D。 et al。 EClinicalMedicine35, 100894 (2021)。
12。 Finkel, Y。 et al。 Nature 594, 240–245 (2021)。
13。 Schubert, K。 et al。 Nature Struct。 Mol。 Biol。27, 959–966 (2020)。
14。 Thoms, M。 et al。 Science 369, 1249–1255 (2020)。
15。 Zhang, K。 et al。 Sci。 Adv。 7, eabe7386 (2021)。
16。 Blanco-Melo, D。 et al。 Cell 181, 1036–1045 (2020)。
17。 Thorne, L。 G。 et al。 Preprint at bioRxiv https://doi.org/10.1101/2021.06.06.446826 (2021)。
18。 Braga, L。 et al。 Nature594, 88–93 (2021)。
19。 Zhang, Z。 et al。 Cell Death Differ。 https://doi.org/10.1038/s41418-021-00782-3 (2021)。
20。 Trimarco, J。 D。 et al。 PLoS Pathog。 17, e1009599 (2021)。
21。 Wolff, G。 et al。 Science369, 1395–1398 (2020)。
22。 Ghosh, S。 et al。 Cell 183, 1520–1535 (2020)。
23。 Hoffmann, M。, Kleine-Weber, H。 & P?hlmann, S。 Mol。 Cell78, 779–784 (2020)。
24。 Mykytyn, A。 Z。 et al。 eLife 10, e64508 (2021)。
25。 Gordon, D。 E。 et al。 Nature 583, 459–468 (2020)。
26。 Li, B。 et al。 Preprint at medRxiv https://doi.org/10.1101/2021.07.07.21260122 (2021)。
原文以How the coronavirus infects cells — and why Delta is so dangerous標題發表在2021年7月28日的《自然》的新聞特寫版塊上
版權聲明:
本文由施普林格·自然上海辦公室負責翻譯。中文內容僅供參考,一切內容以英文原版為準。歡迎轉發至朋友圈,如需轉載,請郵件China@nature.com。未經授權的翻譯是侵權行為,版權方將保留追究法律責任的權利。



“掌”握科技鮮聞 (微信搜索techsina或掃描左側二維碼關注)











